In the last post, we have looked at the Deutsch-Jozsa algorithm that is considered to be the first example of a quantum algorithm that is structurally more efficient than any classical algorithm can probably be. However, the problem solved by the algorithm is rather special. This does, of course, raise the question whether a similar speed-up can be achieved for problems that are more relevant to practical applications.
In this post, we will discuss an algorithm of this type – Grover’s algorithm. Even though the speed-up provided by this algorithm is rather limited (which is of a certain theoretical interest in its own right), the algorithm is interesting due to its very general nature. Roughly speaking, the algorithm is concerned with an unstructured search. We are given a set of N = 2n elements, labeled by the numbers 0 to 2n-1, exactly one of which having a property denoted by P. We can model this property as a binary valued function P on the set 
Grover’s algorithm
Grover’s algorithm presented in [1] proceeds as follows to locate this element. First, we again apply the Hadamard-Walsh operator W to the state 
- Apply a conditional phase shift S, i.e. apply the unique unitary transformation that maps
to
.
- Apply the unitary transformation D called diffusion that we will describe below
Finally, after a defined number of outcomes, we perform a measurement which will collaps the system into one of the states
Before we can proceed, we need to define the matrix D. This matrix is 
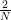
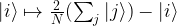
Consequently, we see that
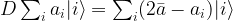
where 
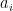
For the sake of completeness, let us also briefly discuss the first transformation employed by the algorithm, the conditional phase shift. We have already seen a similar transformation while studying the Deutsch-Jozsa algorithm. In fact, we have shown in the respective blog post that the circuit displayed below (with the notation slightly changed)
performs the required operation
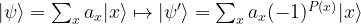
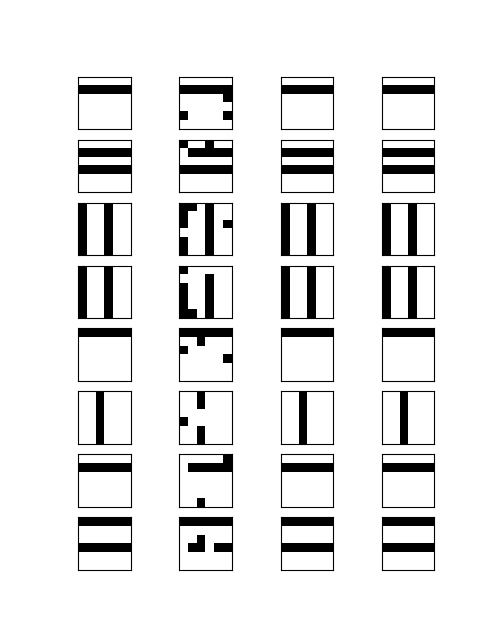
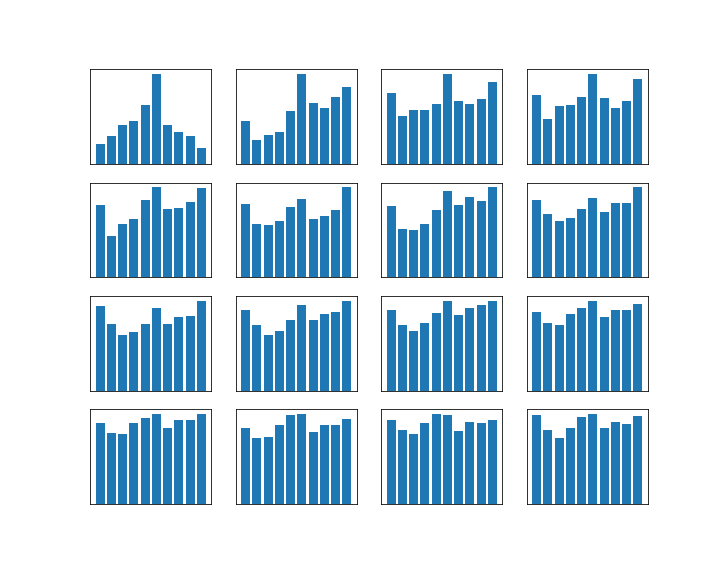
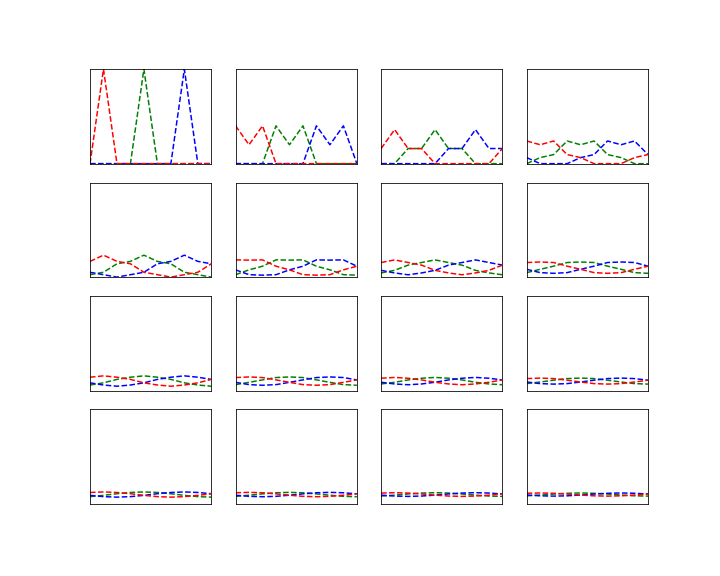
Let us now see how why Grover’s algorithm works. Instead of going through the careful analysis in [1], we will use bar charts to visualize the quantum states (exploiting that all involved matrices are actually real valued).
It is not difficult to simulate the transformation in a simple Python notebook, at least for small values of N. This script performs several iterations of the algorithm and prints the result. The diagrams below show the outcome of this test.
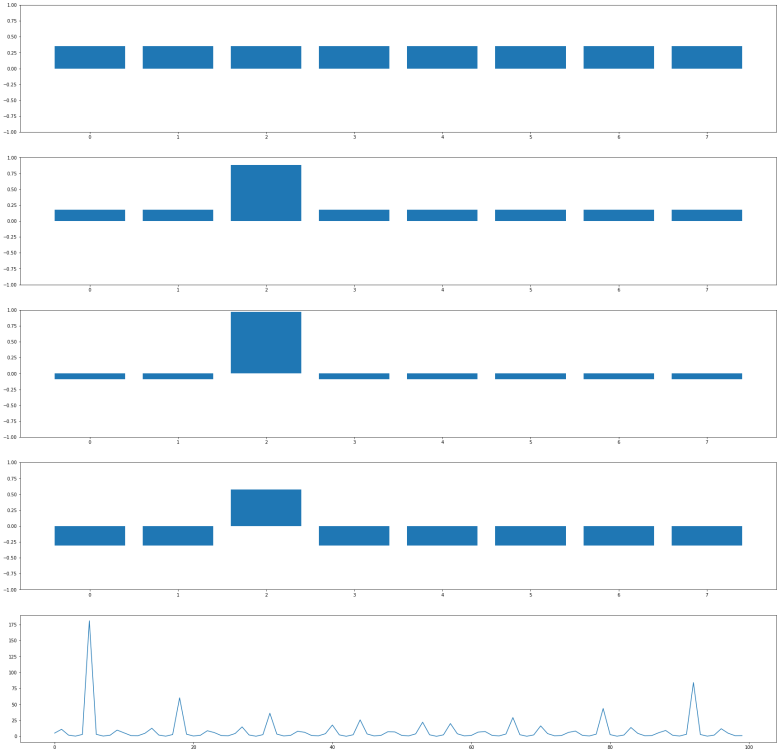
Let us go through the diagrams one by one. The first diagram shows the initial state of the algorithm. I have used 3 qubits, i.e. n = 3 and N = 8. The initial state, after applying the Hadamard-Walsh transform to the zero state, is displayed in the first line. As expected, all amplitudes are equal to 1 over the square root of eight, which is approximately 0.35, i.e. we have a balanced superposition of all states.
We now apply one iteration of the algorithm. First, we apply the conditional phase flip. The element we are looking for is in this case located at x = 2. Thus, the phase flip will leave all basis vectors unchanged except for 
Thus, what really happens in this case is an amplitude amplification – we increase the amplitude of one component of the superposition while decreasing all the others.
The next few lines show the result of repeating these two steps. We see that after the second iteration, almost all of the amplitude is concentrated on the vector
It is interesting to see that when we perform one more iteration, the difference between the amplitude of the solution and the amplitudes of all other components decreases again. Thus the correct choice for the number of iterations is critical to make the algorithm work. In the last line, we have plotted the difference between the amplitude of 
Generalizations and amplitude amplification
In a later paper ([3]), Grover describes a more general setup which is helpful to understand the basic reason why the algorithm works – the amplitude amplification. In this paper, Grover argues that given any unitary transformation U and a target state (in our case, the state representing the solution to the search problem), the probability to meet the target state by applying U to a given initial state can be amplified by a sequence of operations very much to the one considered above. We will not go into details, but present a graphical representation of the algorithm.
So suppose that we are given an n-qubit quantum system and two basis vectors – the vector t representing the target state and an initial state s. In addition, assume we are given a unitary transformation U. The goal is to reach t from s by subsequently applying U itself and a small number of additional gates.
Grover considers the two-dimensional subspace spanned by the vectors s and U-1t. If, within this subspace, we ever manage to reach U-1t, then of course one more application of U will move us into the desired target state.
Now let us consider the transformation
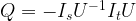
where Ix denotes a conditional phase shift that flips the phase on the vector
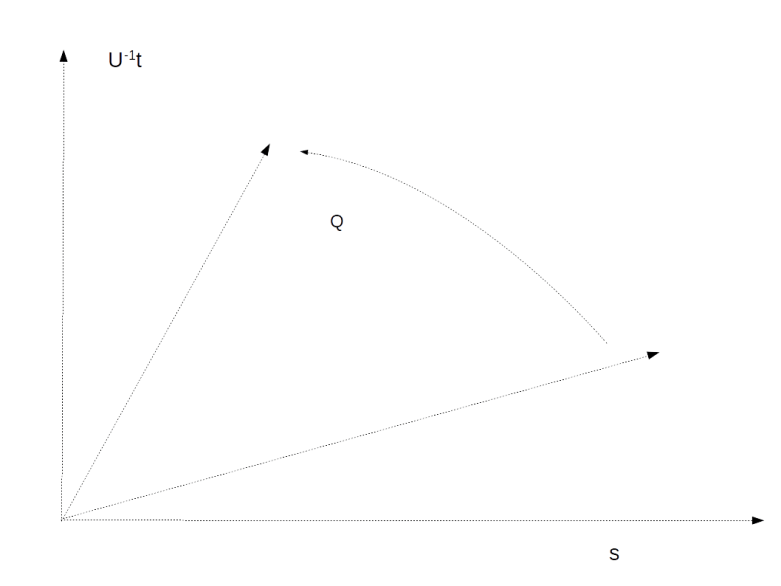
He then proceeds to show that for sufficiently small values of the matrix element Uts, the action of Q on this subspace can approximately be described as a rotation by the angle 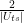

Thus if we can make the first coefficient very small, i.e. if 
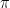
Let us link this description to the version of the Grover algorithm discussed above. In this version, the initial state s is the state 

This can be written as
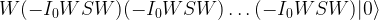
Regrouping this and using the relation D = -W I0 W, we see that this is the same as

Thus the algorithm can equally well be described as applying W once to obtain a balanced superposition and then applying the sequence DS n times, which is the formulation of the algorithm used above. As 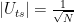
Applications
Grover’s algorithm is highly relevant for theoretical reasons – it applies to a very generic problem and (see the discussions in [1] and [2]) is optimal, in the sense that it provides a quadratic speedup compared to the best classical algorithm that requires O(N) operations, and that this cannot be improved further. Thus Grover’s algorithm provides an interesting example for a problem where a quantum algorithm delivers a significant speedup, but no exponential speedup as we will see it later for Shor’s algorithm.
However, as discussed in detail in section 9.6 of [4], the relevance of the algorithm for practical applications is limited. First, the algorithm applies to an unstructured search, i.e. a search over unstructured data. In most practical applications, we deal with databases that have some sort of structure, and then more efficient search algorithms are known. Second, we have seen that the algorithm requires 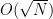
Examples of such problems are brute forces searches as they appear in some crypto-systems. Suppose for instance we are trying to break a message that is encrypted with a symmetric key K, and suppose that we know the first few characters of the original text. We could then try to use an unstructured search over the space of all keys to find a key which matches at least the few characters that we know.
In [5], a more detailed analysis of the complexity in terms of qubits and gates that a quantum computer would have to attack AES-256 is made, arriving at a size of a few thousand logical quantum bits. Given the current ambition level, this does not appear to be completely out of reach. It does, however, not render AES completely unsecure. In fact, as Grover’s algorithm essentially results in a quadratic speedup, a code with a key length of n bits in a pre-quantum world is essentially as secure as the same code with a key length of 2n in a post-quantum world, i.e. roughly speaking, doubling the key length compensates the advantage of quantum computing in this case. This is the reason why the NIST report on post-quantum cryptography still classifies AES as inherently secure assuming increased key sizes.
In addition, the feasibility of a quantum algorithm is not only determined by the number of qubits required, but also by other factors like the depth, i.e. the number of operations required, and the number of quantum gates – and for AES, the estimates in [5] are significant, for instance a depth of more than 2145 for AES-256, which is roughly 1043. Even if we assume a switching time of only 10-12 seconds, we still would require astronomical 1031 seconds, i.e. in the order of 1023 years, to run the algorithm.
Even a much less sophisticated analysis nicely demonstrates the problem behind these numbers – the number of iterations required. Suppose we are dealing with a key length of n bits. Then we know that the algorithm requires

iterations. Taking the decimal logarithm, we see that this is in the order of 100.15*n. Thus, for n = 256, we need in the order of 1038 iterations – a number that makes it obvious that AES-256 can still be considered secure for all practical purposes.
So overall, there is no reason to be overly concerned about serious attacks to AES with sufficiently large keys in the near future. For asymmetric keys, however, we will soon see that the situation is completely different – algorithms like RSA or Elliptic curve cryptography are once and for all broken as soon as large-scale usable quantum computer become reality. This is a consequence of Shor’s algorithm that we will study soon. But first, we need some more preliminaries that we will discuss in the next post, namely quantum Fourier transforms.
References
1. L.K. Grover, A fast quantum mechanical algorithm for database search, Proceedings, 28th Annual ACM Symposium on the Theory of Computing (STOC), May 1996, pages 212-219, available as arXiv:quant-ph/9605043v3
[2] M. Boyer, G. Brassard, P. Høyer, A. Tapp, Tight bounds on quantum searching, arXiv:quant-ph/9605034
3. L.K. Grover, A framework for fast quantum mechanical algorithms, arXiv:quant-ph/9711043
4. E. Rieffel, W. Polak, Quantum computing – a gentle introduction, MIT Press
5. M. Grassl, B. Langenberg, M. Roetteler, R. Steinwandt, Applying Grover’s algorithm to AES: quantum resource estimates, arXiv:1512.04965

 for these two possibilities. In a classical setting, we could then try to encode one bit in the state of such a system, where TRUE might correspond to
for these two possibilities. In a classical setting, we could then try to encode one bit in the state of such a system, where TRUE might correspond to 
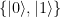 as eigenvectors, with eigenvalues 0 and 1, i.e. it is given by the matrix
as eigenvectors, with eigenvalues 0 and 1, i.e. it is given by the matrix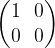

 , we can form the tensor product
, we can form the tensor product 

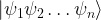
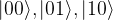 and
and 
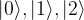 and
and 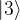
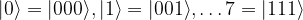
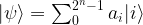
 and
and  , we obtain
, we obtain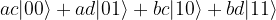
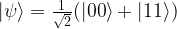
 . Thus this is an example of a state which cannot be written as a tensor product of two single qubit states (but it can be written, of course, as a sum of such states). States that can be decomposed as a product are called separable states, and states that are not separable are called entangled states.
. Thus this is an example of a state which cannot be written as a tensor product of two single qubit states (but it can be written, of course, as a sum of such states). States that can be decomposed as a product are called separable states, and states that are not separable are called entangled states.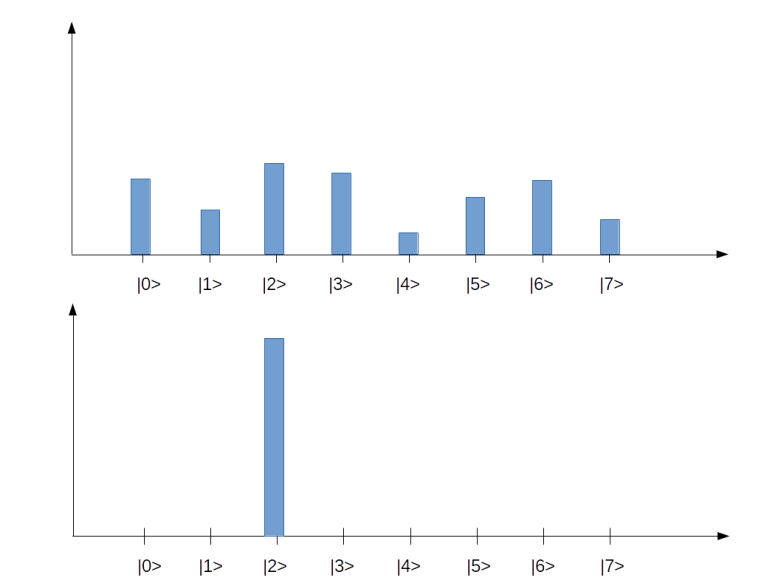
 . In our example, we have eight basis states, corresponding to a quantum system with 3 qubits. The bar represents the amplitude, i.e. the number
. In our example, we have eight basis states, corresponding to a quantum system with 3 qubits. The bar represents the amplitude, i.e. the number  in this case). These states correspond to the eight states of a corresponding classical system with three bits, whereas all states with more than one bar are true superpositions which have no direct equivalent in the classical case. We will later see how similar diagrams can be used to visualize the inner workings of simple quantum algorithms.
in this case). These states correspond to the eight states of a corresponding classical system with three bits, whereas all states with more than one bar are true superpositions which have no direct equivalent in the classical case. We will later see how similar diagrams can be used to visualize the inner workings of simple quantum algorithms.
 , in practice this could be weights, bias terms or any other parameters. To simplify things a bit, we will also assume that the latent variable is finite. Our aim is to maximize the log likelihood, which we can – under these assumptions – express as follows.
, in practice this could be weights, bias terms or any other parameters. To simplify things a bit, we will also assume that the latent variable is finite. Our aim is to maximize the log likelihood, which we can – under these assumptions – express as follows.
 . For that purpose, we introduce a term that is traditionally called Q and defined as follows (all this is a bit abstract, but will become clearer later when we do an example):
. For that purpose, we introduce a term that is traditionally called Q and defined as follows (all this is a bit abstract, but will become clearer later when we do an example):![Q(\Theta'; \Theta) = E \left[ \ln P(x,z | \Theta') | x, \Theta \right]](https://s0.wp.com/latex.php?latex=Q%28%5CTheta%27%3B+%5CTheta%29+%3D+E+%5Cleft%5B++%5Cln+P%28x%2Cz+%7C+%5CTheta%27%29++%7C+x%2C+%5CTheta+%5Cright%5D++&bg=FFFFFF&fg=000&s=1&c=20201002)
 of z. Thus the right hand side is, spelled out
of z. Thus the right hand side is, spelled out![E \left[ \ln P(x,z | \Theta') | x, \Theta \right] = \sum_z \ln P(x,z | \Theta') P(z | x, \Theta)](https://s0.wp.com/latex.php?latex=E+%5Cleft%5B++%5Cln+P%28x%2Cz+%7C+%5CTheta%27%29++%7C+x%2C+%5CTheta+%5Cright%5D+%3D+%5Csum_z+%5Cln+P%28x%2Cz+%7C+%5CTheta%27%29+P%28z+%7C+x%2C+%5CTheta%29++&bg=FFFFFF&fg=000&s=1&c=20201002)
 of parameters such that when passing from
of parameters such that when passing from  to
to  , the value of Q does not decrease, i.e.
, the value of Q does not decrease, i.e.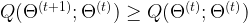

 is maximized. This part of the algorithm is therefore called the maximization step. Then we start over, using
is maximized. This part of the algorithm is therefore called the maximization step. Then we start over, using 
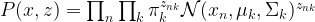

 and
and  . Using this notation, we can now write down the result of calculating the Q function:
. Using this notation, we can now write down the result of calculating the Q function: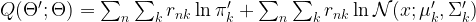
 . For a fixed
. For a fixed  and
and  , and we can now try to maximize this function with respect to these parameters. This calculation is not difficult, but again a bit tiresome (and requires the use of Lagrangian multipliers as there is a constraint on the
, and we can now try to maximize this function with respect to these parameters. This calculation is not difficult, but again a bit tiresome (and requires the use of Lagrangian multipliers as there is a constraint on the 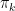 ), and I again refer to
), and I again refer to 
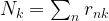


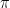 , the covariance matrices
, the covariance matrices  and the means
and the means  – which could, for instance, be chosen randomly. Then, we calculate the responsibilities as above – essentially, this is the expectation step, as it amounts to finding Q.
– which could, for instance, be chosen randomly. Then, we calculate the responsibilities as above – essentially, this is the expectation step, as it amounts to finding Q. 
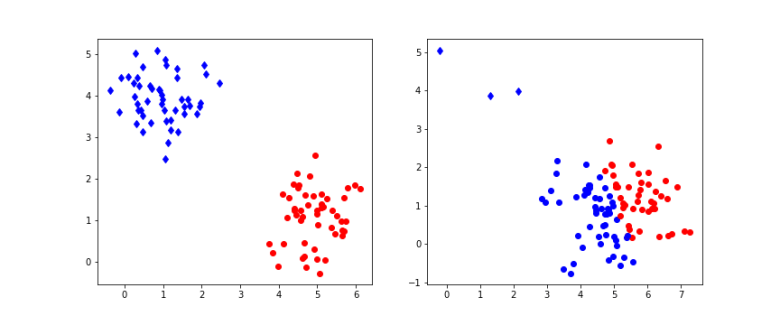
 of points in some euclidian space and a number K of clusters. We want to identify the centre
of points in some euclidian space and a number K of clusters. We want to identify the centre 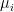 of each cluster and then assign the data points xi to some of the
of each cluster and then assign the data points xi to some of the  , namely to the
, namely to the 

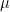 of cluster centers, we can easily minimize Rij by assigning each data point to the cluster whose center is closest to xi. Thus we set
of cluster centers, we can easily minimize Rij by assigning each data point to the cluster whose center is closest to xi. Thus we set

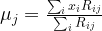

 is a multivariate Gaussian distribution with mean
is a multivariate Gaussian distribution with mean 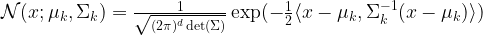
 .
. with the additional constraint that only one of the Zk is allowed to be different from zero. We interpret
with the additional constraint that only one of the Zk is allowed to be different from zero. We interpret 


 is as above and
is as above and
 . As we already have k, this amounts to sampling from the Gaussian distribution
. As we already have k, this amounts to sampling from the Gaussian distribution 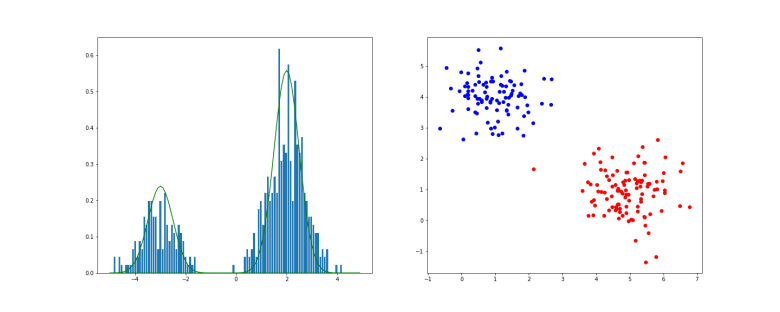

 . Another data point the network could get is
. Another data point the network could get is

 to be a bird is a function of the Xi. In other words, using the language of conditional probabilities,
to be a bird is a function of the Xi. In other words, using the language of conditional probabilities,
 with a vector valued random variable X. The attributes of a sample are described by the feature vector X in some subset of m-dimensional euclidian space, where m is the number of different features. In our example, m=2, as we try to classify animals based on two properties. The result of the classification is described by a random variable Y taking – for the simple case of a binary classification problem – values in
with a vector valued random variable X. The attributes of a sample are described by the feature vector X in some subset of m-dimensional euclidian space, where m is the number of different features. In our example, m=2, as we try to classify animals based on two properties. The result of the classification is described by a random variable Y taking – for the simple case of a binary classification problem – values in 
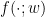 is a function parametrized by some parameter w that we call the weights of the model. The actual value w0 of w is unknown. Based on a sample for X and Y, we then try to fit the model, i.e. we try to find a value for w such that
is a function parametrized by some parameter w that we call the weights of the model. The actual value w0 of w is unknown. Based on a sample for X and Y, we then try to fit the model, i.e. we try to find a value for w such that  models the actual conditional distribution of Y as good as possible. Once the fitting phase is completed, we can then use the model to derive predictions about objects which are not in our initial sample set.
models the actual conditional distribution of Y as good as possible. Once the fitting phase is completed, we can then use the model to derive predictions about objects which are not in our initial sample set.

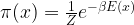
 for a single point in the state space intractable.
for a single point in the state space intractable.

 .
. 

 . We now calculate
. We now calculate 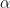 according to the formula above. We then accept the proposal with probability
according to the formula above. We then accept the proposal with probability  . If the proposal is accepted, we set xn+1 = y, otherwise we set xn+1 = xn, i.e. we stay where we are.
. If the proposal is accepted, we set xn+1 = y, otherwise we set xn+1 = xn, i.e. we stay where we are.

 with probability one. This is very similar to a random search for a global maximum – we start at some point x, choose a candidate for a point with higher value of
with probability one. This is very similar to a random search for a global maximum – we start at some point x, choose a candidate for a point with higher value of 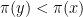 with a non-zero probability. This allows the algorithm to escape a local maximum much better. Intuitively, the algorithm will still try to spend more time in regions with large values of
with a non-zero probability. This allows the algorithm to escape a local maximum much better. Intuitively, the algorithm will still try to spend more time in regions with large values of 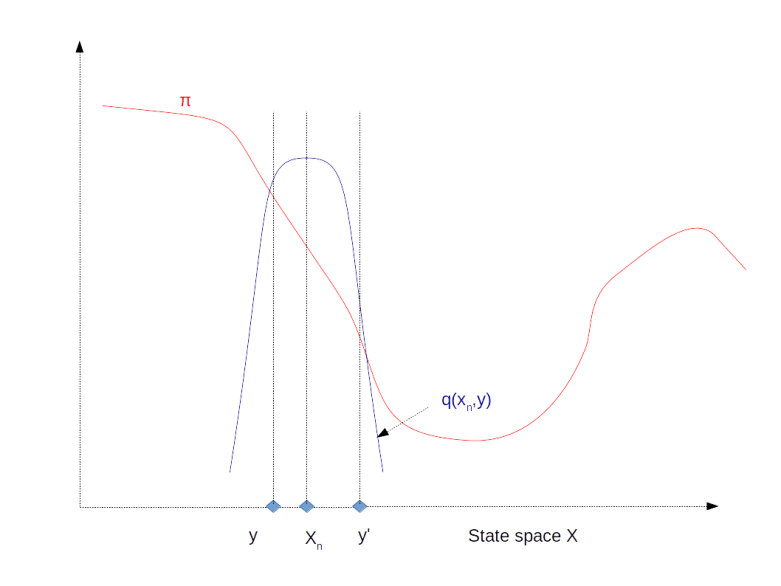
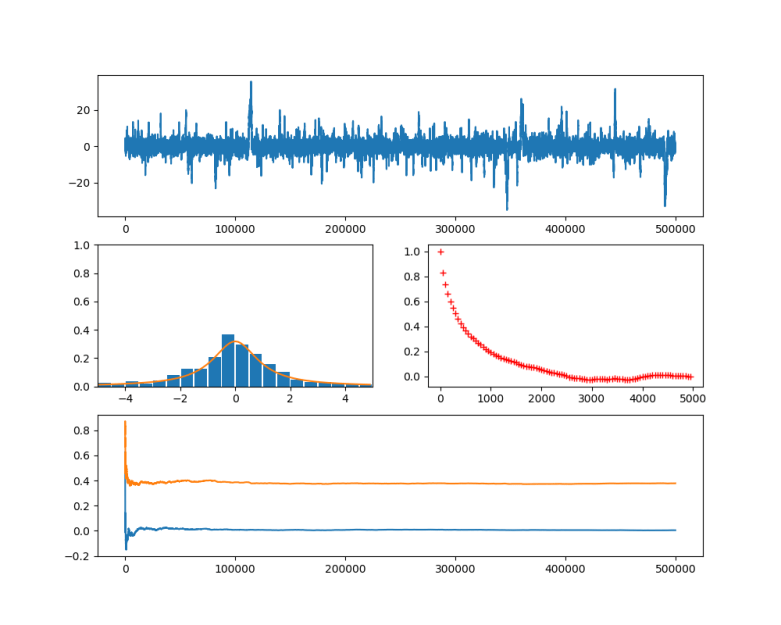
 and
and  ) approximated using the partial sums develops over time. We see that even though we still have huge spikes, the integral remains comparatively stable and converges already after a few thousand iterations. Even if we run the simulation only for 1000 steps, we already get close to the actual values zero (for
) approximated using the partial sums develops over time. We see that even though we still have huge spikes, the integral remains comparatively stable and converges already after a few thousand iterations. Even if we run the simulation only for 1000 steps, we already get close to the actual values zero (for  (for
(for 
 and
and 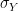 are the standard deviations of X and Y. In our case, given a lag, i.e. a number l less than the length of the chain, we can form two samples, one consisting of the points
are the standard deviations of X and Y. In our case, given a lag, i.e. a number l less than the length of the chain, we can form two samples, one consisting of the points  and the second one consisting of the points of the shifted series
and the second one consisting of the points of the shifted series  . The autocorrelation with lag l is then defined to be the correlation coefficient between these two series. In the diagram, we can see how the autocorrelation depends on the lag. We see that for a large lag, the autocorrelation becomes small, supporting our intuition that the series and the shifted series become independent. However, if we execute several simulation runs, we will also find that in some cases, the convergence of the autocorrelation is very slow, so care needs to be taken when trying to obtain a nearly independent sample from the chain.
. The autocorrelation with lag l is then defined to be the correlation coefficient between these two series. In the diagram, we can see how the autocorrelation depends on the lag. We see that for a large lag, the autocorrelation becomes small, supporting our intuition that the series and the shifted series become independent. However, if we execute several simulation runs, we will also find that in some cases, the convergence of the autocorrelation is very slow, so care needs to be taken when trying to obtain a nearly independent sample from the chain.  , we can use the conditional probability given either x1 or x2 as a proposal distribution. Thus, we first fix x2, and draw a new value for x1 from the conditional probability for x1 given the current value of x2. Then we move to this new coordinate, fix x1, draw from the conditional distribution of x2 given x1 and set the new value of x2 accordingly. It can be shown (see for example
, we can use the conditional probability given either x1 or x2 as a proposal distribution. Thus, we first fix x2, and draw a new value for x1 from the conditional probability for x1 given the current value of x2. Then we move to this new coordinate, fix x1, draw from the conditional distribution of x2 given x1 and set the new value of x2 accordingly. It can be shown (see for example 
 where
where 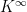 such that
such that
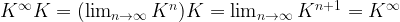
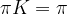 . Interpreting K as transition probabilities, this implies that if the distribution of Xn is
. Interpreting K as transition probabilities, this implies that if the distribution of Xn is  converges to
converges to 
 and the chain converges, but every vector with row sum one is an invariant distribution. This chain is too rigid, because in whatever state we start, we will stay in this state forever. It turns out that in order to ensure uniquess of an invariant distribution, we need a certain property that makes sure that the states can move around freely which is called irreducibility.
and the chain converges, but every vector with row sum one is an invariant distribution. This chain is too rigid, because in whatever state we start, we will stay in this state forever. It turns out that in order to ensure uniquess of an invariant distribution, we need a certain property that makes sure that the states can move around freely which is called irreducibility.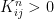 . In other words, given a row index i and a column index j, we can find a power n such that the element of Kn at (i,j) is not zero. It turns out that if a chain is not irreducible, we can split the state space into smaller areas that the chain – once it has entered one of them – does not leave again and on which it is irreducible. So irreducible Markov chains are the buildings blocks of more general Markov chains, and the study of many properties of Markov chains can be reduced to the irreducible case.
. In other words, given a row index i and a column index j, we can find a power n such that the element of Kn at (i,j) is not zero. It turns out that if a chain is not irreducible, we can split the state space into smaller areas that the chain – once it has entered one of them – does not leave again and on which it is irreducible. So irreducible Markov chains are the buildings blocks of more general Markov chains, and the study of many properties of Markov chains can be reduced to the irreducible case.

 -irreducibility. Once we have that, we find two potential behaviours that do not show up in the finite case.
-irreducibility. Once we have that, we find two potential behaviours that do not show up in the finite case.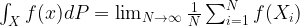
 will, for large n, be approximately identically distributed, namely according to
will, for large n, be approximately identically distributed, namely according to 

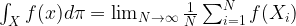
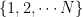 where N is the number of states. We also assume that all our points are measurable, i.e we consider our state space as a discrete probability space.
where N is the number of states. We also assume that all our points are measurable, i.e we consider our state space as a discrete probability space.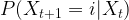
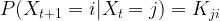
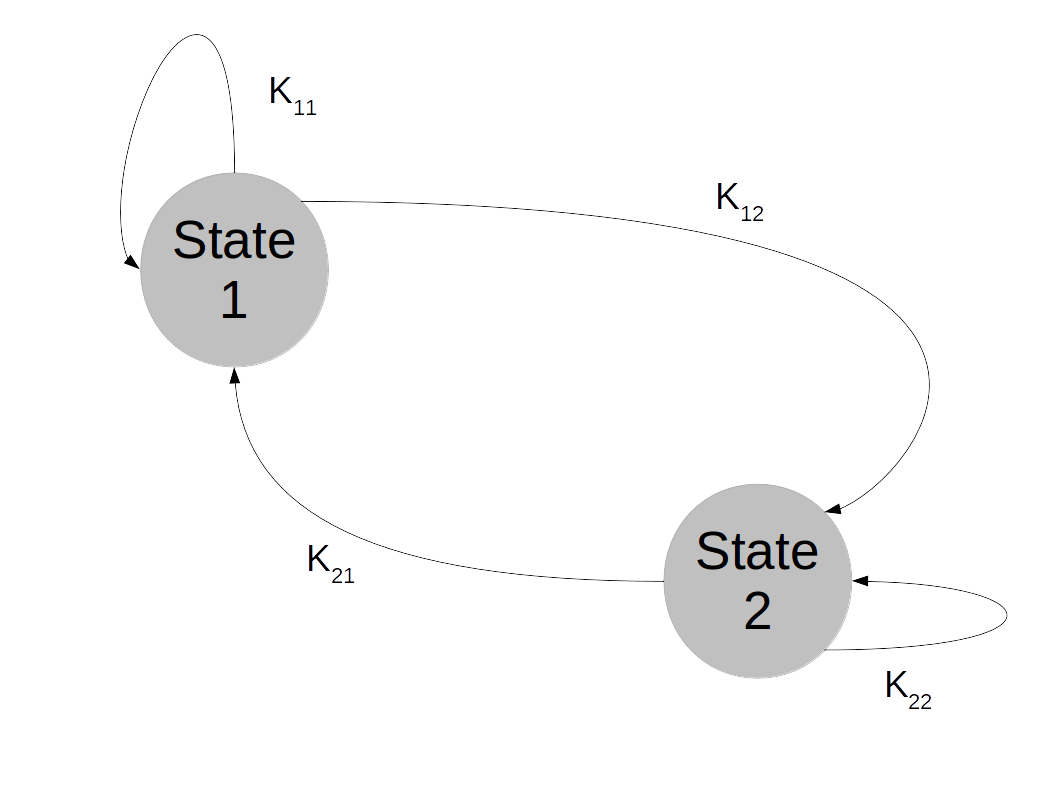
 of random variables taking values in X with the Markov property saying that for all times t, the conditional distribution for Xt+1 given all previous values
of random variables taking values in X with the Markov property saying that for all times t, the conditional distribution for Xt+1 given all previous values 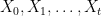 only depends on Xt, i.e.
only depends on Xt, i.e.
 . We can think of the combined values
. We can think of the combined values  for all times t as a history of states or as a random walk in the state space. The Markov property then means that the probability to transition into a next state does not depend on the full history, but only on the current state – Markov chains do not have a memory.
for all times t as a history of states or as a random walk in the state space. The Markov property then means that the probability to transition into a next state does not depend on the full history, but only on the current state – Markov chains do not have a memory.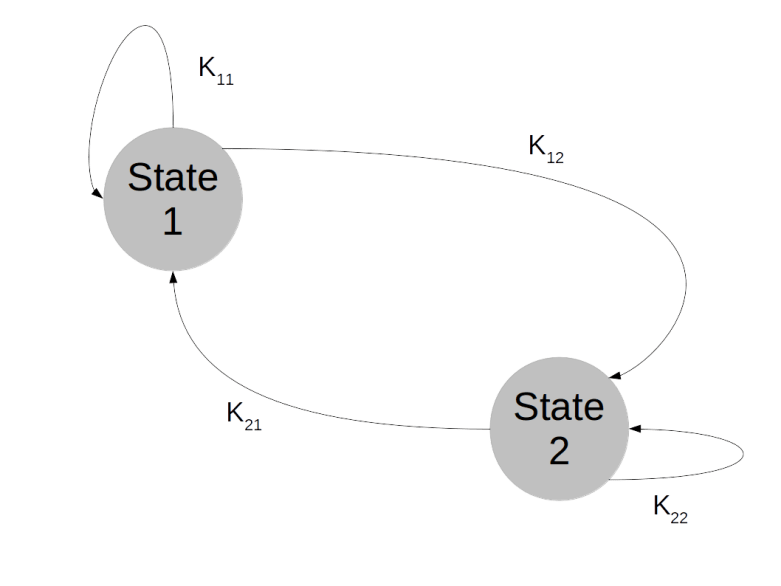



 .
.

 , then the transition matrix is given by a matrix of the form (for instance for N=4)
, then the transition matrix is given by a matrix of the form (for instance for N=4)



 . If we take
. If we take