
So far, I have exclusively been using AWS EC2 when I needed access to a GPU – not because I have carefully compared the available offerings and taken a deliberate decision, but simply because I already had an EC2 account and know the platform.
However, I though it would be interesting to try out other platforms as well. In this post, I will talk a bit about my experience with Paperspace. This provider has several offerings – Core, which is basically an IaaS service, and Gradient, which allows you to access Jupyter notebooks and run jobs in ready-made environments optimized for Machine Learning – and of course I wanted to try this.
It should be noted that some time has passed between trying this out for the first time (roughly in May) and publication of this post in July, so bear with me when some of the details have changed in the meantime – Paperspace is still under development.
First steps
After signing up, you are routed to a page where you can choose between two products – Paperspace Core and Paperspace Gradient. I did choose Gradient (after providing the requested credit card information). The first thing I did try was to bring up a Jupyter notebook.
When you select that option, you have to make two choices. First, Jupyter notebooks are started in Docker containers, and you have to pick one of the available containers. Second, and more important, you have to select a machine – you have a choice between several CPU based and several GPU based models with different fees associated with them.
After a few seconds, your notebook is up and running (with the base account, you can only have one notebook server at any point in time). If you hit “Open”, a new tab will open and you will see the usual Jupyter home screen.
Your notebook folder will be prepopulated with some tutorials. The one I tried first is one of the classical MNIST / CNN tutorials. Unfortunately, when I tried to run it, the kernel died several times in a row – not very encouraging (it worked two days later, and overall there seem to be a few sporadic errors that come and go over time..).
Next, I could not resist the temptation to open a terminal. The Docker image seems to be based on a very basic Ubuntu distribution. I could successfully do an apt-get update && apt-get install git. So you could probably start to download things and work directly from the console – but of course this is not really the idea.
After playing for some time with the notebook, you can – again on the Paperspace notebook console stop your notebook (make sure to do this, you will be charged while the notebook is running). Once the notebook has stopped, you can click on the little arrow to the right of the notebook name, which will give you the option to download any files in the notebook directory that you have created in your session.
Once stopped, there is no way to restart a notebook, but you can clone a notebook which will create a copy of the previous notebook and start that copy, so you can continue to work where you left off. This works, but is a bit tiresome as you have to delete the obsolete copy manually.
Jobs
The next thing I tried is to create a job. For that purpose, you will first have to install the Paperspace CLI which in turn requires node.js and npm. So here is what you need to do on Ubuntu:
$ cd ~ $ apt install nodejs $ apt install npm $ npm install paperspace-node $ sudo ln -s /usr/bin/nodejs /usr/bin/node
This will create a directory node_modules in your home directory and within that directory, a directory .bin. To test the paperspace CLI, you can run a command like
$ ./node_modules/.bin/paperspace --version
Next, switch to an empty directory and in that directory, run
$ ~/node_modules/.bin/paperspace project init
This will initiate a new paperspace project, i.e. it will create a subdirectory .ps_project containing a JSON configuration file. Next, you need an API key that you can get on your Paperspace home page. The API key is an authentication token that is used by the API – store that number in a safe place.
Once we have that token, it is time to start our first job.
~/node_modules/.bin/paperspace jobs create --container Test-Container --command "nvidia-smi" --apiKey "xxxxx" --machineType K80
where xxxxx needs to be replaced by your API key. Instead of providing your API key with every command, you can also run
$ ~/node_modules/.bin/paperspace login
which will add your credentials to a file in the .paperspace directory in your home directory.
Essentially, what happens when you run a job is that the local directory and all its subdirectories will be zipped into a file, a container will be set up on a Paperspace server, the content of the ZIP file will be extracted into this container and the command that you have specified will execute.
You can now get a list of your processes and their status either on the Paperspace console, where you also have immediate access to the log output, or from the command line using
$ ~/node_modules/.bin/paperspace jobs list
At this point, I was again a bit disappointed – the job appears to be running and is even displayed in the web console, but when it completes, I get an error “503 – Service unavailable” and no log output is provided. I raised a request with the support, and roughly 2 hours later the submission suddenly worked – I have not yet found out whether the support has really done anything or whether a part of the infrastructure was really down at this point in time.
As a temporary workaround, I managed to run a job by redirecting error output and standard output to a file. For instance, to run the script KMeans.py, I did use
$ ~/node_modules/.bin/paperspace jobs create --command "export MPLBACKEND=AGG ; python KMeans.py > /artifacts/log 2>&1" --machineType C2
Once the job is complete, you can download whatever it has added to the directory /artifacts using
$ ~/node_modules/.bin/paperspace jobs artifactsGet --jobId "js26y3pi6jk056"
where the job ID is the ID of the job and will be displayed by the create command. Finally, the command jobs destroy --jobId=... can be used to delete a job after execution.
So far, I have to admit that I am not so happy with what I have seen. I hit upon several issues in the standard setup, and when playing around, I found that it can take a long time for a job to be scheduled on a GPU (depending very much on the machine type – my impression was that for machine types for which Paperspace uses GCP, like K80 or P100, your job will run quickly, but for other types like GPU+ it can take a long time). In addition, as everything is running in a container, the initial steps in job can be time consuming. TensorFlow, for instance, is known to take longer when it is started the first time, and in a fresh container, every time is a first time, so you will see a significant startup time. This gets worse if you need to download data sets, as this will have to be repeated with every new run. It is apparently not yet possible to mount a permanent volume into your container to avoid this or to reuse a stopped container (update: as of July, Paperspace has announced that the /storage directory is a persistent storage available across notebooks and jobs, but I have not yet tried this).
But maybe this is a premature judgement, and I decided that I will still continue to try it out. In one of the next posts, I will present some more advanced features and in particular the Python API that Paperspace offers.

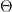 , in practice this could be weights, bias terms or any other parameters. To simplify things a bit, we will also assume that the latent variable is finite. Our aim is to maximize the log likelihood, which we can – under these assumptions – express as follows.
, in practice this could be weights, bias terms or any other parameters. To simplify things a bit, we will also assume that the latent variable is finite. Our aim is to maximize the log likelihood, which we can – under these assumptions – express as follows.
 . For that purpose, we introduce a term that is traditionally called Q and defined as follows (all this is a bit abstract, but will become clearer later when we do an example):
. For that purpose, we introduce a term that is traditionally called Q and defined as follows (all this is a bit abstract, but will become clearer later when we do an example):![Q(\Theta'; \Theta) = E \left[ \ln P(x,z | \Theta') | x, \Theta \right]](https://s0.wp.com/latex.php?latex=Q%28%5CTheta%27%3B+%5CTheta%29+%3D+E+%5Cleft%5B++%5Cln+P%28x%2Cz+%7C+%5CTheta%27%29++%7C+x%2C+%5CTheta+%5Cright%5D++&bg=FFFFFF&fg=000&s=1&c=20201002)
 of z. Thus the right hand side is, spelled out
of z. Thus the right hand side is, spelled out![E \left[ \ln P(x,z | \Theta') | x, \Theta \right] = \sum_z \ln P(x,z | \Theta') P(z | x, \Theta)](https://s0.wp.com/latex.php?latex=E+%5Cleft%5B++%5Cln+P%28x%2Cz+%7C+%5CTheta%27%29++%7C+x%2C+%5CTheta+%5Cright%5D+%3D+%5Csum_z+%5Cln+P%28x%2Cz+%7C+%5CTheta%27%29+P%28z+%7C+x%2C+%5CTheta%29++&bg=FFFFFF&fg=000&s=1&c=20201002)
 of parameters such that when passing from
of parameters such that when passing from  to
to  , the value of Q does not decrease, i.e.
, the value of Q does not decrease, i.e.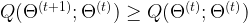

 is maximized. This part of the algorithm is therefore called the maximization step. Then we start over, using
is maximized. This part of the algorithm is therefore called the maximization step. Then we start over, using 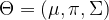
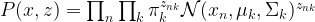

 and
and  . Using this notation, we can now write down the result of calculating the Q function:
. Using this notation, we can now write down the result of calculating the Q function: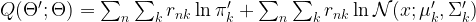
 . For a fixed
. For a fixed  and
and  , and we can now try to maximize this function with respect to these parameters. This calculation is not difficult, but again a bit tiresome (and requires the use of Lagrangian multipliers as there is a constraint on the
, and we can now try to maximize this function with respect to these parameters. This calculation is not difficult, but again a bit tiresome (and requires the use of Lagrangian multipliers as there is a constraint on the 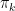 ), and I again refer to
), and I again refer to 
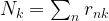


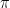 , the covariance matrices
, the covariance matrices  and the means
and the means  – which could, for instance, be chosen randomly. Then, we calculate the responsibilities as above – essentially, this is the expectation step, as it amounts to finding Q.
– which could, for instance, be chosen randomly. Then, we calculate the responsibilities as above – essentially, this is the expectation step, as it amounts to finding Q. 
 of points in some euclidian space and a number K of clusters. We want to identify the centre
of points in some euclidian space and a number K of clusters. We want to identify the centre 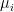 of each cluster and then assign the data points xi to some of the
of each cluster and then assign the data points xi to some of the  , namely to the
, namely to the 

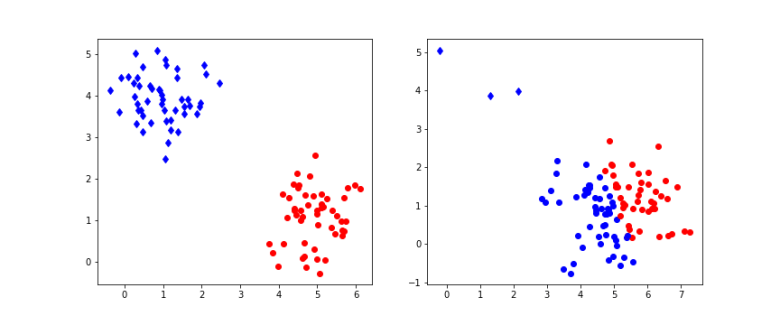
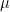 of cluster centers, we can easily minimize Rij by assigning each data point to the cluster whose center is closest to xi. Thus we set
of cluster centers, we can easily minimize Rij by assigning each data point to the cluster whose center is closest to xi. Thus we set

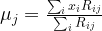

 is a multivariate Gaussian distribution with mean
is a multivariate Gaussian distribution with mean 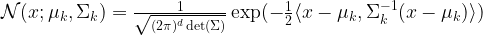
 .
. with the additional constraint that only one of the Zk is allowed to be different from zero. We interpret
with the additional constraint that only one of the Zk is allowed to be different from zero. We interpret 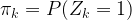


 is as above and
is as above and
 . As we already have k, this amounts to sampling from the Gaussian distribution
. As we already have k, this amounts to sampling from the Gaussian distribution 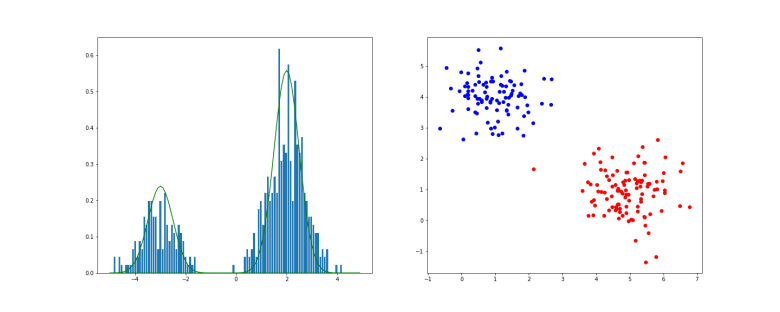


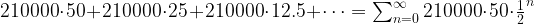
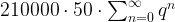
 . This series converges, and its value is
. This series converges, and its value is
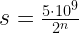
 for n, we obtain
for n, we obtain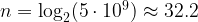
 .
.
