
In this post, we will investigate the Metropolis-Hastings algorithm, which is still one of the most popular algorithms in the field of Markov chain Monte Carlo methods, even though its first appearence (see [1]) happened in 1953, more than 60 years in the past. It does for instance appear on the CiSe top ten list of the most important algorithms of the 20th century (I got this and the link from this post on WordPress).
Before we get into the algorithm, let us once more state the problem that the algorithm is trying to solve. Suppose you are given a probability distribution 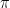

of the quantity f. Thus to make a prediction that can be verified or falsified by an observation, you will have to calculate integrals of this type.
Now, in practice, this can be very hard. One issue is that in order to naively calculate the integral, you would have to transverse the entire state space, which is not feasible for most realistic problems as this tends to be a very high dimensional space. Closely related to this is a second problem. Remember, for instance, that a typical distribution like the Boltzmann distribution is given by
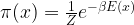
The term in the numerator is comparatively easy to calculate. However, the term in the denominator is the partition function, and is itself an integral over the state space! This makes even the calculation of 
But there is hope – even though calculating the values of

for large values of N.
But how do we construct a Markov chain that converges to a given distribution? The Metropolis Hastings approach to solve this works as follows.
The first thing that we do is to choose a proposal density q on our state space X, i.e. a measurable function

such that for each x, 
Then q defines a Markov chain, where the probability to transition into a measurable set A being at a point x is given by the integral

Of course this is not yet the Markov chain that we want – it has nothing to do with

This number is called the acceptance probability, and, as promised, it only contains ratios of probabilities, so that factors like the partition function cancel and do not have to be computed.
The Metropolis Hastings algorithm now proceeds as follows. We start with some arbitrary point x0. When the chain has arrived at xn, we first draw a candidate y for the next location from the proposal distribution 
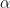

Clearly, the xn are samples from a Markov chain, as the position at step xn only depends on the position at step xn-1. But is still appears to be a bit mysterious why this should work. To shed light on this, let us consider a case where the expressions above simplify a bit. So let us assume that the proposal density q is symmetric, i.e. that

This is the original Metropolis algorithm as proposed in [1]. If we also assume that

Thus we accept the proposal if 
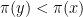
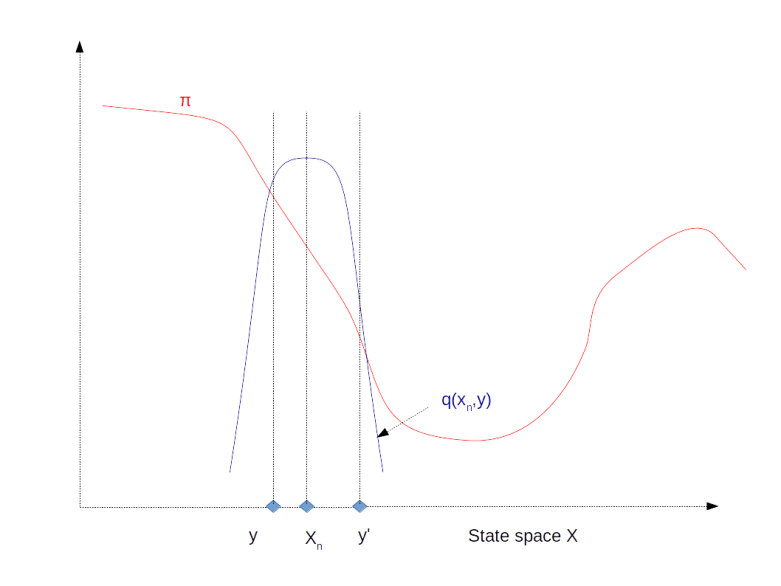
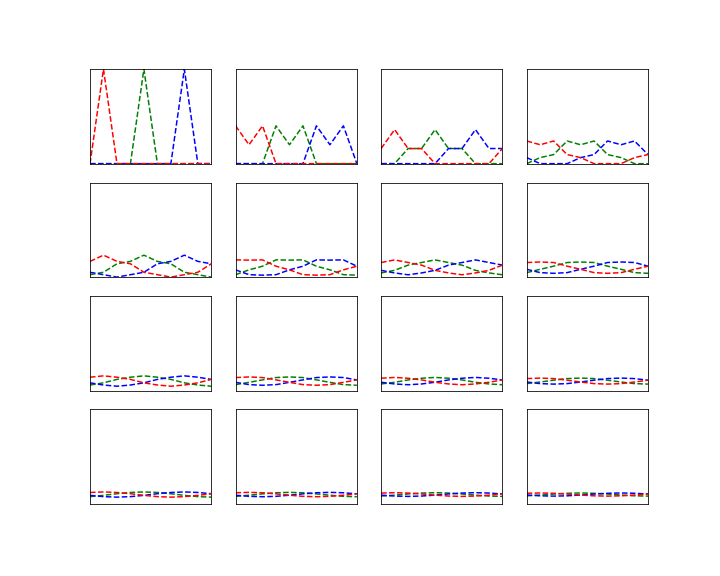
The image above illustrates this procedure. The red graph displays the distribution
In this form, the algorithm is extremely easy to implement. All we need is a function propose that creates the next proposal, and a function p that calculates the value of the probability density
import numpy as np
chain = []
X = 0
chain.append(X)
for n in range(args.steps):
Y = propose(X)
U = np.random.uniform()
alpha = p(Y) / p(X)
if (U <= alpha):
X = Y
chain.append(X)
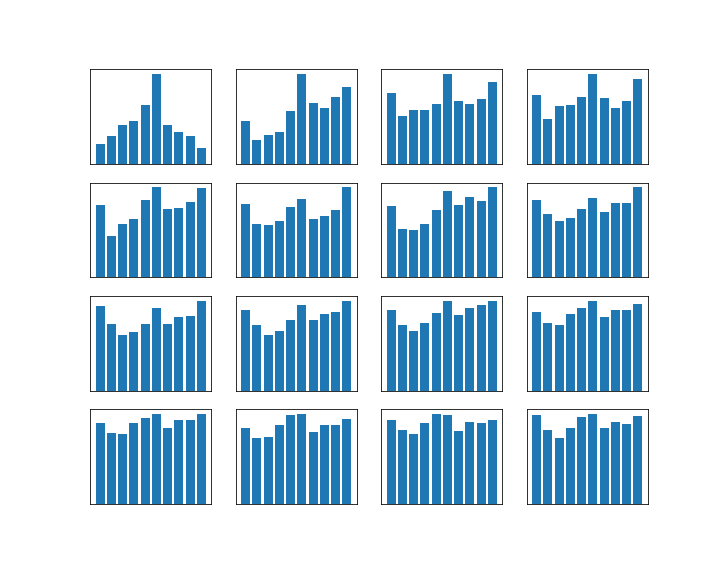
In the diagram below, this algorithm has been applied to a Cauchy distribution with mode zero and scale one, using a normal distribution with mean x and standard deviation 0.5 as a proposal for the next location. The chain was calculated for 500.000 steps. The diagram in the upper part shows the values of the chain during the simulation.
Then the first 100.000 steps were discarded and considered as "burn-in" time for the chain to stabilize. Out of the remaining 400.000 sample points, points where chosen with a distance of 500 time steps to obtain a sample which is approximately independent and identically distributed. This is called subsampling and typically not necessary for Monte Carlo integration (see [2], chapter 1 for a short discussion of the need of subsampling), but is done here for the sake of illustration. The resulting subsample is plotted as a histogramm in the lower left corner of the diagram. The yellow line is the actual probability density.
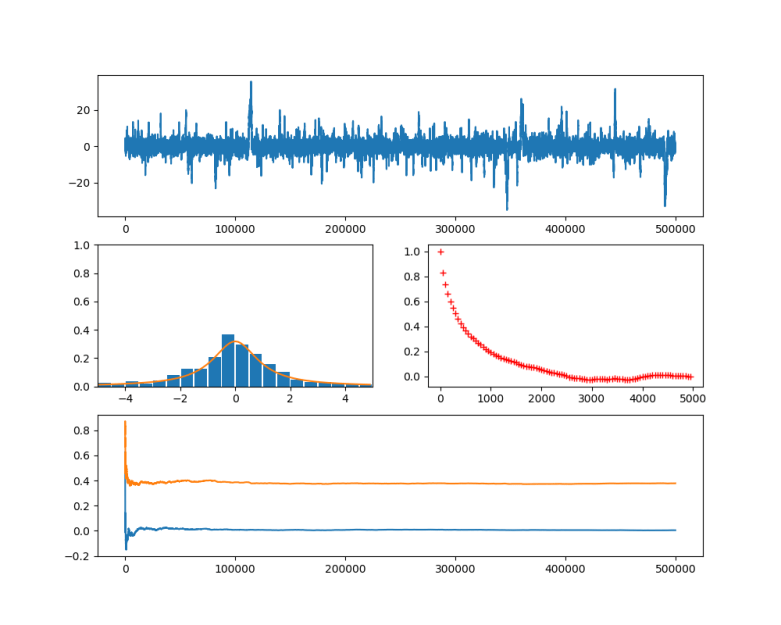
We see that after a few thousand steps, the chain converges, but continues to have spikes. However, the sampled distribution is very close to the sample generated by the Python standard method (which is to take the quotient of two independent samples from a standard normal distribution).
In the diagram at the bottom, I have displayed how the integral of two functions (


scipy.integrate.quad integration routine).
In the second diagram in the middle row, I have plotted the autocorrelation versus the lag, as an indicator for the failure of the sample points to be independent. Recall that for two samples X and Y, the Pearson correlation coefficient is the number

where 
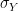


In practice, the autocorrelation is probably not a good measure for the convergence of a Markov chain. It is important to keep in mind that obtaining an independent sample is not the point of the Markov chain – the real point is that even though the sample is autocorrelated, we can approximate expectation values fairly well. However, I have included the autocorrelation here for the sake of illustration.
This form of proposal distributions is not the only one that is commonly used. Another choice that appears often is called an independence sampler. Here the proposal distribution is chosen to be independent of the current location x of the chain. This gives us an algorithm that resembles the importance sampling method and also shares some of the difficulties associated with it – in my notes on Markov chain Monte Carlo methods, I have included a short discussion and a few examples. These notes also contain further references and a short discussion of why and when the Markov chain underlying a Metropolis-Hastings sampler converges.
Other variants of the algorithm work by updating – in a high-dimensional space – either only one variable at a time or entire blocks of variables that are known to be independent.
Finally, if we are dealing with a state space that can be split as a product 
Monte Carlo sampling methods are a broad field, and even though this has already been a long post, we have only scratched the surface. I invite you to consult some of the references below and / or my notes for more details. As always, you will also find the sample code on GitHub and might want to play with this to reproduce the examples above and see how different settings impact the result.
In a certain sense, this post is the last post in the series on restricted Boltzmann machines, as it provides (at least some of) the mathematical background behind the Gibbs sampling approach that we used there. Boltzmann machines are examples for stochastic neuronal networks that can be applied to unsupervised learning, i.e. the allow a model to learn from a sample distribution without the need for labeled data. In the next few posts on machine learning, I will take a closer look at some other algorithms that can be used for unsupervised learning.
References
1. N. Metropolis,A.W. Rosenbluth, M.N. Rosenbluth, A.H. Teller, E. Teller, Equation of state calculation by fast computing machines, J. Chem. Phys. Vol. 21, No. 6 (1953), pp. 1087-1092
2. S. Brooks, A. Gelman, C.L. Jones,X.L. Meng (ed.), Handbook of Markov chain Monte Carlo, Chapman Hall / CRC Press, Boca Raton 2011
3. W.K. Hastings, Monte Carlo sampling methods using Markov chains and their applications, Biometrika, Vol. 57 No. 1 (1970), pp. 97-109
R.M. Neal, Probabilistic inference using Markov chain Monte Carlo methods, Technical Report CRG-TR-93-1, Department of Computer Science, University of Toronto, 1993
5. C.P. Robert, G. Casella, Monte Carlo Statistical Methods,
Springer, New York 1999

 where
where 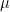 is the initial distribution. So if Kn converges, we can expect the distribution of Xn to converge as well. If there is a matrix
is the initial distribution. So if Kn converges, we can expect the distribution of Xn to converge as well. If there is a matrix 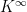 such that
such that
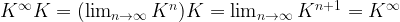
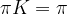 . Interpreting K as transition probabilities, this implies that if the distribution of Xn is
. Interpreting K as transition probabilities, this implies that if the distribution of Xn is  converges to
converges to 
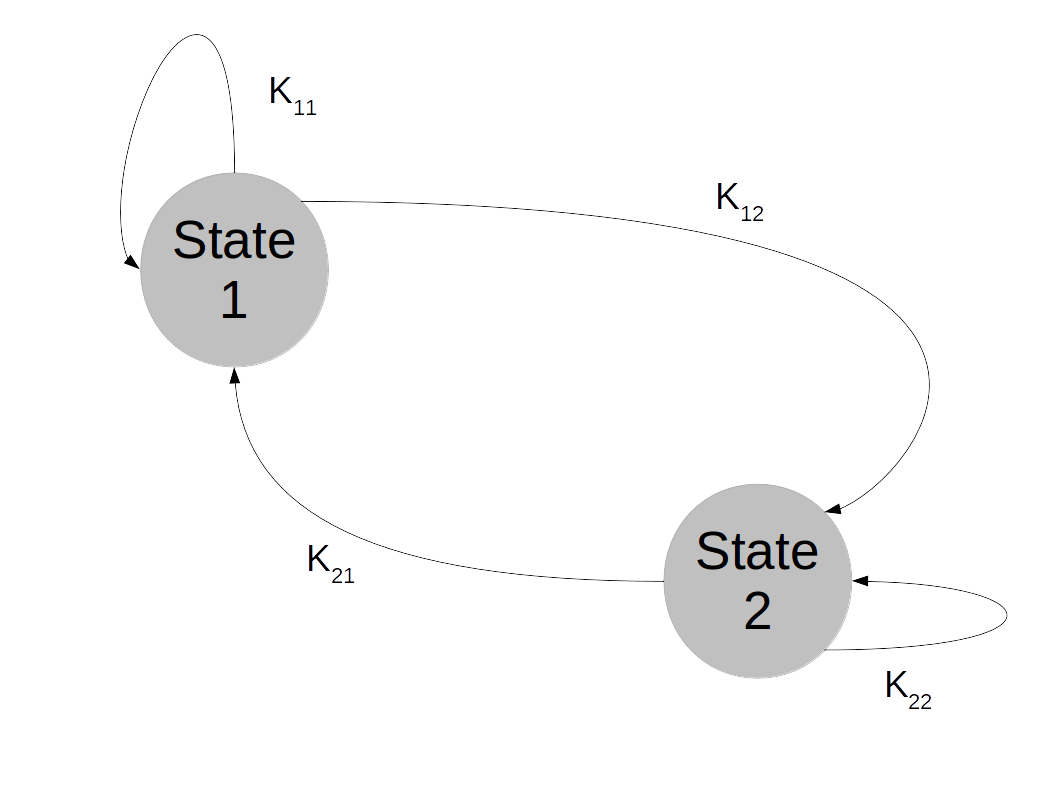
 and the chain converges, but every vector with row sum one is an invariant distribution. This chain is too rigid, because in whatever state we start, we will stay in this state forever. It turns out that in order to ensure uniquess of an invariant distribution, we need a certain property that makes sure that the states can move around freely which is called irreducibility.
and the chain converges, but every vector with row sum one is an invariant distribution. This chain is too rigid, because in whatever state we start, we will stay in this state forever. It turns out that in order to ensure uniquess of an invariant distribution, we need a certain property that makes sure that the states can move around freely which is called irreducibility.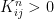 . In other words, given a row index i and a column index j, we can find a power n such that the element of Kn at (i,j) is not zero. It turns out that if a chain is not irreducible, we can split the state space into smaller areas that the chain – once it has entered one of them – does not leave again and on which it is irreducible. So irreducible Markov chains are the buildings blocks of more general Markov chains, and the study of many properties of Markov chains can be reduced to the irreducible case.
. In other words, given a row index i and a column index j, we can find a power n such that the element of Kn at (i,j) is not zero. It turns out that if a chain is not irreducible, we can split the state space into smaller areas that the chain – once it has entered one of them – does not leave again and on which it is irreducible. So irreducible Markov chains are the buildings blocks of more general Markov chains, and the study of many properties of Markov chains can be reduced to the irreducible case.

 -irreducibility. Once we have that, we find two potential behaviours that do not show up in the finite case.
-irreducibility. Once we have that, we find two potential behaviours that do not show up in the finite case.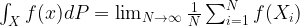
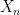 will, for large n, be approximately identically distributed, namely according to
will, for large n, be approximately identically distributed, namely according to 

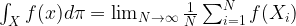
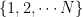 where N is the number of states. We also assume that all our points are measurable, i.e we consider our state space as a discrete probability space.
where N is the number of states. We also assume that all our points are measurable, i.e we consider our state space as a discrete probability space.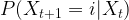
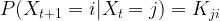
 of random variables taking values in X with the Markov property saying that for all times t, the conditional distribution for Xt+1 given all previous values
of random variables taking values in X with the Markov property saying that for all times t, the conditional distribution for Xt+1 given all previous values 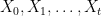 only depends on Xt, i.e.
only depends on Xt, i.e.
 . We can think of the combined values
. We can think of the combined values  for all times t as a history of states or as a random walk in the state space. The Markov property then means that the probability to transition into a next state does not depend on the full history, but only on the current state – Markov chains do not have a memory.
for all times t as a history of states or as a random walk in the state space. The Markov property then means that the probability to transition into a next state does not depend on the full history, but only on the current state – Markov chains do not have a memory.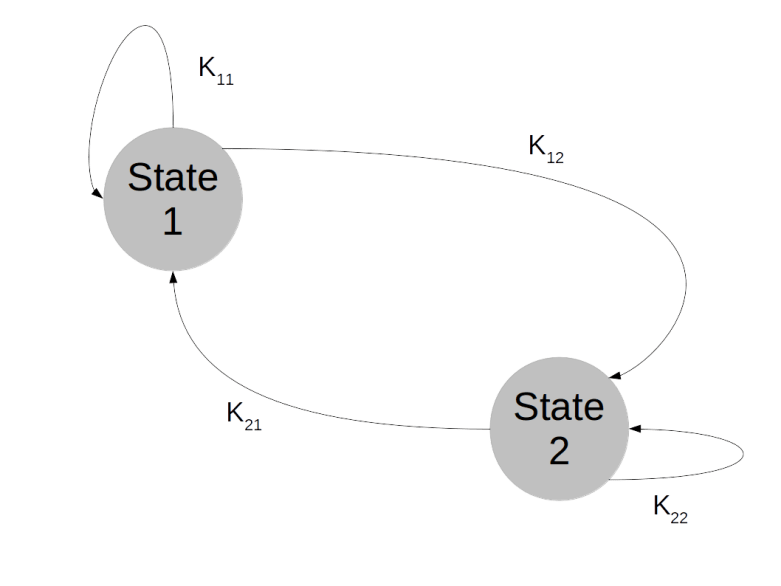
 . What is the probability to be in state j after one step? Of course we can write
. What is the probability to be in state j after one step? Of course we can write


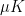 .
.

 , then the transition matrix is given by a matrix of the form (for instance for N=4)
, then the transition matrix is given by a matrix of the form (for instance for N=4)



 . If we take
. If we take 

